Autore principale: Santi, Valentina
Pubblicazione: Firenze : Regione Toscana, 2004
Tipo di risorsa: testo, Livello bibliografico: monografia, Lingua: ita, Paese: IT

La malattia di Alzheimer-Perusini, detta anche morbo di Alzheimer, demenza presenile di tipo Alzheimer, demenza degenerativa primaria di tipo Alzheimer o semplicemente Alzheimer, è la forma più comune di demenza degenerativa progressivamente invalidante con esordio prevalentemente in età presenile (oltre i 65 anni). Nel DSM-5 viene nominata come disturbo neurocognitivo maggiore o lieve dovuto a malattia di Alzheimer (331.0). Si stima che circa il 50-70% dei casi di demenza sia dovuta a tale condizione, mentre il 10-20% a demenza vascolare. Il sintomo precoce più frequente è la difficoltà nel ricordare eventi recenti. Con l'avanzare dell'età possiamo avere sintomi come: afasia, disorientamento, cambiamenti repentini di umore, depressione, incapacità di prendersi cura di sé, problemi nel comportamento. Ciò porta il soggetto inevitabilmente a isolarsi nei confronti della società e della famiglia. A poco a poco, le capacità mentali basilari vengono perse. Anche se la velocità di progressione può variare, l'aspettativa media di vita dopo la diagnosi è dai tre ai nove anni.La patologia è stata descritta per la prima volta nel 1906, dallo psichiatra e neuropatologo tedesco Alois Alzheimer. Nel 2006 vi erano 26,6 milioni di malati in tutto il mondo e si stima che ne sarà affetta 1 persona su 85 a livello mondiale entro il 2050.La causa e la progressione della malattia di Alzheimer (Alzheimer's Disease, AD) non sono ancora ben compresi. La ricerca indica che la malattia è strettamente associata a placche amiloidi e ammassi neurofibrillari riscontrati nel cervello, ma non è nota la causa prima di tale degenerazione. Attualmente i trattamenti terapeutici utilizzati offrono piccoli benefici sintomatici e possono parzialmente rallentare il decorso della patologia; anche se sono stati condotti oltre 500 studi clinici per l'identificazione di un possibile trattamento per l'Alzheimer, non sono ancora stati identificati trattamenti che ne arrestino o invertano il decorso. Circa il 70% del rischio si ritiene sia genetico con molti geni solitamente coinvolti. Altri fattori di rischio includono: traumi, depressione o ipertensione. Il processo della malattia è associata a placche amiloidi che si formano nel SNC.Una diagnosi probabile è basata sulla progressione della malattia, test cognitivi con imaging medico e gli esami del sangue per escludere altre possibili cause. I sintomi iniziali sono spesso scambiati per normale invecchiamento. È necessaria la biopsia del tessuto cerebrale per una diagnosi definitiva. L'esercizio mentale e fisico possono diminuire il rischio di AD. Non esistono farmaci o integratori che scientificamente possano diminuire il rischio di AD.A livello preventivo, sono state proposte diverse modificazioni degli stili di vita personali come potenziali fattori protettivi nei confronti della patologia, ma non vi sono adeguate prove di una correlazione certa tra queste raccomandazioni e la riduzione effettiva della degenerazione. Stimolazione mentale, esercizio fisico e una dieta equilibrata sono state proposte sia come modalità di possibile prevenzione, sia come modalità complementari di gestione della malattia.. La sua ampia e crescente diffusione nella popolazione, la limitata e, comunque, non risolutiva efficacia delle terapie disponibili e le enormi risorse necessarie per la sua gestione (sociali, emotive, organizzative ed economiche), che ricadono in gran parte sui familiari dei malati, la rendono una delle patologie a più grave impatto sociale del mondo.Anche se il decorso clinico della malattia di Alzheimer è in parte specifico per ogni individuo, la patologia causa diversi sintomi comuni alla maggior parte dei pazienti. I primi sintomi osservabili sono spesso erroneamente considerati problematiche "legate all'età", o manifestazioni di stress. Nelle prime fasi, il sintomo più comune è l'incapacità di acquisire nuovi ricordi e la difficoltà nel ricordare eventi osservati recentemente. Quando si ipotizza la presenza di una possibile malattia di Alzheimer, la diagnosi viene di solito confermata tramite specifiche valutazioni comportamentali e test cognitivi, spesso seguiti dall'imaging a risonanza magneticaCon l'avanzare della malattia, il quadro clinico può prevedere confusione, irritabilità e aggressività, sbalzi di umore, difficoltà nel linguaggio, perdita della memoria a breve e lungo termine e progressive disfunzioni sensoriali.Poiché per la malattia di Alzheimer non sono attualmente disponibili terapie risolutive e il suo decorso è progressivo, la gestione dei bisogni dei pazienti diviene essenziale. Spesso è il coniuge o un parente stretto (caregiver) a prendersi in carico il malato, compito che comporta notevoli difficoltà e oneri. Chi si occupa del paziente può sperimentare pesanti carichi personali che coinvolgono aspetti sociali, psicologici, fisici ed economici.
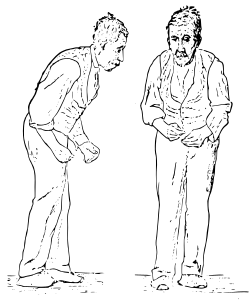
La malattia di Parkinson, sovente definita come morbo di Parkinson, Parkinson, parkinsonismo idiopatico, parkinsonismo primario, sindrome ipocinetica rigida o paralisi agitante è una malattia neurodegenerativa. I sintomi motori tipici della condizione sono il risultato della morte delle cellule che sintetizzano e rilasciano la dopamina. Tali cellule si trovano nella substantia nigra, una regione del mesencefalo. La causa che porta alla loro morte è sconosciuta. All'esordio della malattia i sintomi più evidenti sono legati al movimento, ed includono tremori, rigidità, lentezza nei movimenti e difficoltà a camminare. In seguito possono insorgere problemi cognitivi e comportamentali, con la demenza che si verifica a volte nelle fasi avanzate, ad eccezione dei casi più precoci di demenza da corpi di Lewy, associati a prognosi più grave. La malattia di Parkinson è più comune negli anziani; la maggior parte dei casi si verifica dopo i 50 anni. I sintomi motori principali sono comunemente chiamati parkinsonismo. La condizione è spesso definita come una sindrome idiopatica, anche se alcuni casi atipici hanno un'origine genetica. Molti fattori di rischio e fattori protettivi sono stati indagati: ad esempio, l'aumento del rischio di contrarre la malattia nelle persone esposte ad idrocarburi, solventi e pesticidi. La patologia è caratterizzata dall'accumulo di una proteina, chiamata α-sinucleina, in inclusioni denominate corpi di Lewy nei neuroni e dall'insufficiente formazione di dopamina. La distribuzione anatomica dei corpi di Lewy è spesso direttamente correlata all'espressione e al grado dei sintomi clinici di ciascun individuo. La diagnosi nei casi tipici si basa principalmente sui sintomi, con indagini di neuroimaging come conferma. I moderni trattamenti sono efficaci per gestire i sintomi motori precoci della malattia, grazie all'uso di agonisti della dopamina e del levodopa. Col progredire della malattia, i neuroni dopaminergici continuano a diminuire di numero, e questi farmaci diventano inefficaci nel trattamento della sintomatologia e, allo stesso tempo, producono una complicanza, la discinesia, caratterizzata da movimenti involontari. Una corretta alimentazione e alcune forme di riabilitazione hanno dimostrato una certa efficacia nell'alleviare i sintomi. La chirurgia e la stimolazione cerebrale profonda vengono utilizzate per ridurre i sintomi motori come ultima risorsa, nei casi più gravi in cui i farmaci risultano inefficaci. La malattia prende il nome dal medico inglese James Parkinson, che pubblicò la prima descrizione dettagliata nel suo trattato An Essay on the Shaking Palsy nel 1817.Diverse importanti organizzazioni promuovono la ricerca e il miglioramento della qualità della vita delle persone affette dalla malattia. Alcuni pazienti famosi, come l'attore Michael J. Fox, il pugile Muhammad Ali, papa Giovanni Paolo II, i cantanti Alex Band, Peter Hofmann, Bruno Lauzi e il giornalista Vincenzo Mollica, hanno fatto crescere la consapevolezza della malattia nella società.

La malattia di Huntington, o corea di Huntington (pronuncia: corèa o còrea), è una malattia genetica neurodegenerativa che colpisce la coordinazione muscolare e porta a un declino cognitivo e a problemi psichiatrici. Esordisce tipicamente durante la mezza età; è la più frequente malattia a causa genetica nei quadri clinici neurologici con movimenti involontari anomali (che prendono il nome di corea). È molto più comune nelle persone di discendenza europea occidentale rispetto a chi è di origine asiatica o africana. La malattia è causata da una mutazione autosomica dominante in una delle due copie (alleli) di un gene codificante una proteina chiamata huntingtina, il che significa che ogni figlio di una persona affetta ha una probabilità del 50% di ereditare la condizione. La base genetica della malattia è stata scoperta nel 1993 grazie a una ricerca internazionale guidata dalla Hereditary Disease Foundation. I sintomi fisici della malattia possono incominciare a qualsiasi età, ma più frequentemente tra i 35 e i 44 anni. I sintomi della malattia possono variare tra gli individui e anche tra i membri colpiti della stessa famiglia, ma di solito la loro progressione può essere predetta. I primi sintomi sono spesso sottili problemi di umore o cognitivi a cui segue una generale mancanza di coordinazione e un'andatura instabile. Con l'avanzare della malattia i movimenti non coordinati del corpo diventano sempre più evidenti e sono accompagnati da un calo delle capacità mentali e problemi comportamentali e psichiatrici. Le complicanze, come la polmonite, le malattie cardiache e i danni fisici da cadute, riducono l'aspettativa di vita a circa 20 anni a partire dall'esordio dei sintomi. Non esiste una cura per la condizione e un'assistenza a tempo pieno diventa necessaria nelle fasi più avanzate della malattia. I trattamenti sono solo farmacologici e non possono alleviare molti dei suoi numerosi sintomi.

La paralisi sopranucleare progressiva (PSP) o sindrome di Steele-Richardson-Olszewski è una malattia neurodegenerativa descritta per la prima volta nel 1964. La neurodegenerazione comporta atrofia a livello del mesencefalo e di altre strutture cerebrali tra cui il nucleo subtalamico, il globus pallidus, i nuclei del ponte e la sostanza nera. Il nome della malattia descrive la localizzazione sopranucleare (con riferimento ai nuclei oculomotori del mesencefalo) delle alterazioni patologiche. Il termine 'nucleo' si riferisce al gruppo di neuroni da cui hanno origine i nervi cranici oculomotori che connettono mesencefalo e muscoli estrinseci dell'occhio. Mentre una paralisi dei nervi oculomotori può essere causata da lesioni dei nervi stessi o da lesioni a livello dei nuclei, nel caso della PSP la causa è localizzata nel cervello, al di sopra di quelle strutture. Il quadro clinico porta a sintomi tra cui perdita di equilibrio, rallentamento dei movimenti, difficoltà a muovere gli occhi e demenza.Il quadro istologico è caratterizzato dalla formazione di ammassi neurofibrillari all'interno dei neuroni e depositi patologici nelle cellule gliali. I depositi contengono la proteina tau in stato fosforilato, mentre normalmente questa proteina si trova associata ai microtubuli del citoscheletro. La PSP viene perciò classificata anche come taupatia. Rientra inoltre in un gruppo di disturbi neurologici classificati come "Parkinson-Plus" o "Parkinson atipico" con sintomi che ricordano la malattia di Parkinson, mentre altre caratteristiche sono ben diverse.La PSP è rara con circa 3 casi per 100.000 abitanti ed è alla base di circa il 5% dei disturbi con sintomi parkinsoniani come acinesia e rigidità. La durata media della malattia è di circa 5-10 anni e l'età media all'esordio è di circa 65 anni. I maschi hanno leggermente più probabilità di essere colpiti rispetto alle donne. Non è stata trovata alcuna associazione tra PSP e qualsiasi razza, posizione o occupazione particolare. Nelle prime fasi può essere difficilmente distinguibile dalla Malattia di Parkinson e tra i primi sintomi e la diagnosi definitiva decorre mediamente un tempo di tre anni.
Alcune catalogazioni sono state accorpate perché sembrano descrivere la stessa edizione. Per visualizzare i dettagli di ciascuna, clicca sul numero di record
Record aggiornato il: 2025-09-23T02:14:38.255Z

