
La malattia di Alzheimer-Perusini, detta anche morbo di Alzheimer, demenza presenile di tipo Alzheimer, demenza degenerativa primaria di tipo Alzheimer o semplicemente Alzheimer, è la forma più comune di demenza degenerativa progressivamente invalidante con esordio prevalentemente in età presenile (oltre i 65 anni). Nel DSM-5 viene nominata come disturbo neurocognitivo maggiore o lieve dovuto a malattia di Alzheimer (331.0). Si stima che circa il 50-70% dei casi di demenza sia dovuta a tale condizione, mentre il 10-20% a demenza vascolare. Il sintomo precoce più frequente è la difficoltà nel ricordare eventi recenti. Con l'avanzare dell'età possiamo avere sintomi come: afasia, disorientamento, cambiamenti repentini di umore, depressione, incapacità di prendersi cura di sé, problemi nel comportamento. Ciò porta il soggetto inevitabilmente a isolarsi nei confronti della società e della famiglia. A poco a poco, le capacità mentali basilari vengono perse. Anche se la velocità di progressione può variare, l'aspettativa media di vita dopo la diagnosi è dai tre ai nove anni.La patologia è stata descritta per la prima volta nel 1906, dallo psichiatra e neuropatologo tedesco Alois Alzheimer. Nel 2006 vi erano 26,6 milioni di malati in tutto il mondo e si stima che ne sarà affetta 1 persona su 85 a livello mondiale entro il 2050.La causa e la progressione della malattia di Alzheimer (Alzheimer's Disease, AD) non sono ancora ben compresi. La ricerca indica che la malattia è strettamente associata a placche amiloidi e ammassi neurofibrillari riscontrati nel cervello, ma non è nota la causa prima di tale degenerazione. Attualmente i trattamenti terapeutici utilizzati offrono piccoli benefici sintomatici e possono parzialmente rallentare il decorso della patologia; anche se sono stati condotti oltre 500 studi clinici per l'identificazione di un possibile trattamento per l'Alzheimer, non sono ancora stati identificati trattamenti che ne arrestino o invertano il decorso. Circa il 70% del rischio si ritiene sia genetico con molti geni solitamente coinvolti. Altri fattori di rischio includono: traumi, depressione o ipertensione. Il processo della malattia è associata a placche amiloidi che si formano nel SNC.Una diagnosi probabile è basata sulla progressione della malattia, test cognitivi con imaging medico e gli esami del sangue per escludere altre possibili cause. I sintomi iniziali sono spesso scambiati per normale invecchiamento. È necessaria la biopsia del tessuto cerebrale per una diagnosi definitiva. L'esercizio mentale e fisico possono diminuire il rischio di AD. Non esistono farmaci o integratori che scientificamente possano diminuire il rischio di AD.A livello preventivo, sono state proposte diverse modificazioni degli stili di vita personali come potenziali fattori protettivi nei confronti della patologia, ma non vi sono adeguate prove di una correlazione certa tra queste raccomandazioni e la riduzione effettiva della degenerazione. Stimolazione mentale, esercizio fisico e una dieta equilibrata sono state proposte sia come modalità di possibile prevenzione, sia come modalità complementari di gestione della malattia.. La sua ampia e crescente diffusione nella popolazione, la limitata e, comunque, non risolutiva efficacia delle terapie disponibili e le enormi risorse necessarie per la sua gestione (sociali, emotive, organizzative ed economiche), che ricadono in gran parte sui familiari dei malati, la rendono una delle patologie a più grave impatto sociale del mondo.Anche se il decorso clinico della malattia di Alzheimer è in parte specifico per ogni individuo, la patologia causa diversi sintomi comuni alla maggior parte dei pazienti. I primi sintomi osservabili sono spesso erroneamente considerati problematiche "legate all'età", o manifestazioni di stress. Nelle prime fasi, il sintomo più comune è l'incapacità di acquisire nuovi ricordi e la difficoltà nel ricordare eventi osservati recentemente. Quando si ipotizza la presenza di una possibile malattia di Alzheimer, la diagnosi viene di solito confermata tramite specifiche valutazioni comportamentali e test cognitivi, spesso seguiti dall'imaging a risonanza magneticaCon l'avanzare della malattia, il quadro clinico può prevedere confusione, irritabilità e aggressività, sbalzi di umore, difficoltà nel linguaggio, perdita della memoria a breve e lungo termine e progressive disfunzioni sensoriali.Poiché per la malattia di Alzheimer non sono attualmente disponibili terapie risolutive e il suo decorso è progressivo, la gestione dei bisogni dei pazienti diviene essenziale. Spesso è il coniuge o un parente stretto (caregiver) a prendersi in carico il malato, compito che comporta notevoli difficoltà e oneri. Chi si occupa del paziente può sperimentare pesanti carichi personali che coinvolgono aspetti sociali, psicologici, fisici ed economici.
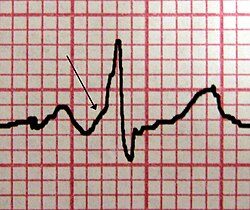
La sindrome di Wolff Parkinson White (WPW) è una malattia congenita da anomala conduzione cardiaca; è caratterizzata dalla presenza di uno o più fasci atrio-ventricolari accessori, che possono dare origine a episodi di tachicardia sporadica. La prevalenza media è stimata in 1/450 e quindi la sindrome non è considerata una malattia rara. La sindrome di WPW colpisce soprattutto maschi (70% dei casi) in giovane età. Durante la vita fetale, gli atri vengono collegati ai ventricoli da numerosi fasci che scompaiono dopo la nascita, fatta eccezione per uno, il fascio di His. Tuttavia, in alcuni pazienti possono persistere anche altri fasci. Queste vie accessorie raggirano il nodo atrio-ventricolare e causano una pre-eccitazione ventricolare e, in alcuni casi, episodi di tachicardia, che possono interrompersi spontaneamente oppure necessitano di un trattamento farmacologico. La sindrome di WPW può essere sporadica o familiare. La forma familiare è difficilmente identificabile, in quanto le vie accessorie non sono sempre attive. Mutazioni del gene PRKAG2 sono state associate a forme familiari della sindrome di WPW. La diagnosi viene spesso posta in base al riscontro dei disturbi del ritmo. Oltre a essi, l'ECG può mostrare tre segni: la mancanza della via lenta atrio-ventricolare (intervallo PR corto, < 0,12 secondi), una pre-eccitazione ventricolare (onda delta all'inizio del complesso QRS), una stimolazione anormale dei ventricoli (complesso QRS più largo> 0,12 secondi tra l'inizio dell'onda delta e la fine di QRS). Queste anomalie dell'ECG corrispondono a un fascio anomalo direttamente collegato agli atri e ai ventricoli. Se il fascio collega l'atrio al fascio di His, l'ECG rileva un intervallo PR corto (< 0,12 secondi), con un complesso QRS normale. Si tratta di una variante della sindrome di WPW, che prende il nome di sindrome di Lown-Ganong-Levine.

Nei primati la mano è l'organo prensile che si trova all'estremità distale dell'arto superiore, collegato a questo tramite il polso. Comprende cinque dita, che costituiscono la parte più predisposta al senso tattile. La mano è il primo strumento del genere umano, nell'Homo sapiens è anche un mezzo di espressione quando aiuta la parola o la sostituisce tramite il linguaggio dei segni.

La malattia di Dupuytren è una patologia a carico della mano caratterizzata dalla flessione progressiva e permanente di uno o più dita, ed è fra tutte le forme di deformità della mano la più comune.
L'algologia, o terapia antalgica, detta anche terapia del dolore o medicina del dolore, consiste nell'approccio terapeutico e scientifico al trattamento del dolore. Il dolore rende spesso il soggetto inabile sia da un punto di vista fisico sia emotivo. Il dolore acuto relativo a un trauma fisico è spesso reversibile naturalmente. Il dolore cronico, invece, generalmente è causato da condizioni solitamente difficili da trattare. Talvolta i neurotrasmettitori continuano a inviare la sensazione del dolore anche quando la causa scatenante non esiste più; per esempio un paziente a cui è stato amputato un arto può provare dolore riferito all'arto mancante (sindrome dell'arto fantasma). Una applicazione dell'algologia è nei malati neoplastici.

